
How to Build an AI ETF Portfolio That Covers the Full Stack
3 hrs ago

The most dangerous assumption in small-cap biotech investing is that all early-stage companies carry the same risk profile. They do not. A subset of biotechnology companies pursues abbreviated regulatory pathways by reformulating existing, well-characterised drugs into new delivery formats rather than synthesising novel molecules from scratch. This approach compresses timelines, lowers development costs, and substantially reduces the safety unknowns that make traditional Phase 1 programmes so binary. Yet many investors apply the same blunt risk discount to these companies as they do to novel drug developers, potentially mispricing the opportunity. What follows uses Patrys (ASX: PAB) and its injectable quetiapine programme for ICU delirium as a concrete case study to explain how drug reformulation works as a biotech investment strategy, what makes it structurally different from standard development, and how finance-oriented investors can evaluate companies pursuing these abbreviated pathways.
ICU delirium affects an estimated 30-70% of intensive care patients. It is not a minor behavioural symptom. It is a serious acute neurological condition that increases mortality, extends hospital stays, and imposes substantial costs on healthcare systems worldwide. Despite that prevalence, delirium has been historically treated as secondary to the conditions that brought patients into critical care.
The scale of the problem is matched only by the absence of a solution. Three characteristics define the commercial gap:
Market sizing: Patrys CEO Dr. Samantha South has consistently cited a US$2 billion annual serviceable addressable market for an approved injectable delirium therapy in hospital and ICU settings.
Certain EU member states permit restricted use of a single antipsychotic for delirium, but that drug carries a notable side effect burden, leaving significant prescribing friction. The result is a large, underserved clinical population and a commercial opening for whoever provides an on-label option first.
Drug reformulation, in plain terms, means converting an existing, approved oral drug into a new delivery format, in this case intravenous, rather than synthesising an entirely new molecule. The underlying drug is already well characterised. Its safety profile is documented across years of clinical use. What changes is how the drug reaches the patient’s bloodstream.
This distinction matters because of how it interacts with regulatory pathways. The FDA’s 505(b)(2) pathway allows applicants to reference existing safety and efficacy data for the underlying molecule. The regulatory burden shifts from proving the drug works from scratch to demonstrating that the new delivery format achieves comparable plasma concentrations without introducing new safety signals. The bridging study required can be conducted in small numbers of subjects over a shorter duration than conventional Phase 1 trials.
The FDA’s 505(b)(2) guidance specifies the criteria under which an applicant may reference previously approved safety and efficacy data, meaning the regulatory burden for a reformulation programme centres on demonstrating bioequivalence rather than rebuilding a full clinical package from first principles.
| Dimension | Novel drug development | Reformulation (505(b)(2)) | Patrys RLS-2201 |
|---|---|---|---|
| Typical timeline | 15-20 years | Materially shorter | First-in-human targeted 2H 2026 |
| Safety unknowns | High (new molecule) | Reduced (known molecule) | Quetiapine safety well documented |
| Phase 1 study size | Large cohort required | Small bridging study | Small-number PK equivalence study |
| Regulatory data burden | Full safety and efficacy package | References existing data | Leverages quetiapine literature |
Timeline compression translates directly into investment risk terms. A reformulation programme consumes less capital before reaching a value inflection event, raising the probability of hitting a readout without requiring a dilutive capital raise. The binary event structure centres on a bridging study, a pharmacokinetic comparison against a known effective dose, rather than on a large Phase 2/3 efficacy outcome where the drug’s fundamental mechanism is being tested for the first time. For investors accustomed to pricing early-stage biotech as a single category, the distinction is material.
Probability-weighted ASX biotech valuation frameworks produce materially different intrinsic estimates depending on which risk categories are assigned higher probabilities of success, and for reformulation programmes the key adjustment is the reduction in Phase 1 safety unknowns rather than any change to efficacy probability, a distinction that standard probability-of-success models applied to novel drug candidates will systematically underweight.
The clinical rationale for IV quetiapine in ICU delirium starts not with pharmacology but with the practical reality of the intensive care ward. Delirious patients are frequently agitated, intubated, or sedated. Persuading them to swallow an oral tablet is, in most cases, clinically unrealistic.
Dr. Samantha South, Patrys CEO, has noted that persuading a delirious ICU patient to swallow a tablet is “virtually impossible,” whereas IV administration is straightforward in a setting where patients already have IV lines in place.
Intravenous delivery addresses this problem while offering three distinct clinical advantages:
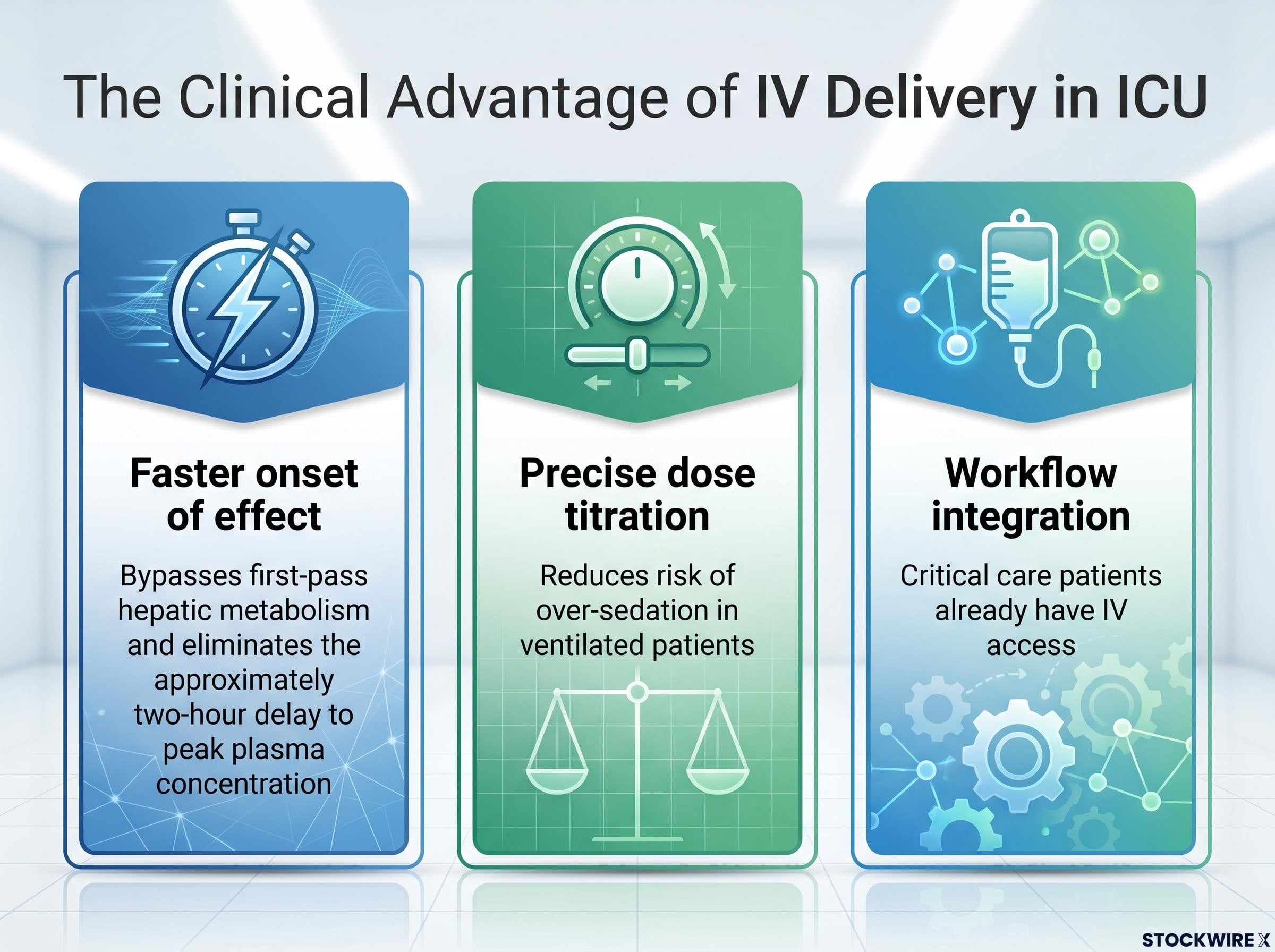
The bridging study itself is designed to demonstrate that IV-delivered quetiapine achieves plasma concentrations comparable to the known effective oral dose, without new adverse signals from the change in delivery method. The programme, designated RLS-2201 (also referred to as RLS-2202 in some company and analyst materials), targets first-in-human clinical trial entry in the second half of 2026. Manufacturing and process validation are underway at BioCina’s sterile facility in Perth. The bridging study is expected to run approximately 3-6 months from initiation, with completion potentially extending into early 2027 depending on start date and recruitment pace.
The RLS-2201 manufacturing progress at BioCina, including engineering batch production and the formal engagement of Facet Life Sciences to progress the 505(b)(2) regulatory strategy, represents the operational foundation that must be in place before first-in-human entry can proceed on the 2H 2026 timeline.
In 2014, Mallinckrodt acquired Cadence Pharmaceuticals for approximately US$1.4 billion. Cadence’s lead product, Ofirmev, was an intravenous reformulation of acetaminophen, an established oral analgesic, developed for hospital post-operative pain management.
The dollar figure is notable, but the structural logic behind the transaction is more instructive. Three dimensions of the Cadence precedent align with the reformulation model:
This precedent is an industry-level data point, not a Patrys valuation guide. The therapeutic areas differ (post-operative pain versus ICU delirium), as do development stages. What the transaction demonstrates is that the exit pathway for this asset class exists and has been executed at scale. That is a separate and important question from whether Patrys will execute successfully.
Reformulation materially reduces certain categories of development risk. The underlying molecule’s safety profile is already documented. The study required for regulatory submission is smaller, shorter, and narrower in scope than a conventional Phase 1 programme. The timeline to a first readout is compressed, reducing the window of capital consumption before a value inflection event.
Those reductions are real. They are also incomplete.
The injectable formulation must still demonstrate acceptable safety in its own right. Management has stated explicitly that reformulation removes much of the unknown safety risk associated with a new chemical entity but does not eliminate clinical risk entirely. Timeline slippage from manufacturing or regulatory factors remains possible. The bridging study readout, while narrower in scope, is still a binary event for the programme. Funding and liquidity risk, a structural feature of ASX-listed junior biotechs, persists regardless of development pathway. A shorter timeline reduces but does not eliminate the probability of a dilutive capital raise.
Investors modelling cash runway for ASX junior biotechs alongside their development timelines should review our full explainer on Australia’s proposed R&D tax cap, which examines the 10-year refundability limit, the support gap it creates for programmes whose commercialisation timelines exceed that window, and the four portfolio variables investors should stress-test before the proposed 2028 commencement date.
| Risk category | Status in reformulation programme |
|---|---|
| Unknown molecular safety | Materially reduced |
| Phase 1 study scope | Materially reduced |
| Timeline to first readout | Materially reduced |
| Bridging study binary event | Persists |
| Funding and dilution risk | Partially mitigated |
| Single-programme concentration | Partially mitigated |
The deoxymab antibody platform, originally licensed from Yale University, provides longer-horizon optionality alongside the nearer-term CNS reformulation programme. Deoxymabs are cell-penetrating antibodies capable of crossing the blood-brain barrier to target intracellular disease mechanisms, including neuroinflammation and cancer. This is not an imminent near-term catalyst. It is a structural diversifier designed to reduce the all-or-nothing profile common in single-asset junior biotechs. The key value inflection window for Patrys remains the 12-month period surrounding 2026 clinical milestones, specifically the pharmacokinetic equivalence data and absence of new safety signals from the initial bridging study readout.
The Patrys case study illustrates a set of analytical criteria that transfer to any small-cap ASX company pursuing a drug reformulation strategy. Five evaluation questions structure the framework:
LTR Pharma’s SPONTAN programme offers a directly comparable example of 505(b)(2) delivery reformulation in action: the same underlying molecule (vardenafil), a new administration route (nasal spray instead of oral tablet), and a primary endpoint focused on pharmacokinetic onset rather than de novo efficacy, compressing the bridging study scope in the same way Patrys targets with RLS-2201.

Primary source verification matters. Programme naming inconsistencies (such as the RLS-2201/RLS-2202 discrepancy in Patrys’ own materials) are a concrete reminder to verify disclosed timelines and regulatory milestone claims against actual ASX filings rather than relying on summary analyst or media materials.
Drug reformulation is a legitimate, structurally distinct investment approach that reduces specific categories of development risk. It is not a risk-free path. It demands the same rigour applied to any early-stage biotech evaluation.
The Patrys case illustrates the model in concrete terms: a US$2 billion addressable market with no approved treatment, an IV reformulation of a well-characterised antipsychotic, a 505(b)(2) bridging study targeting first-in-human entry in 2H 2026, and an industry precedent (Cadence/Mallinckrodt) demonstrating the exit pathway at scale.
The ASX hosts a range of companies pursuing abbreviated development pathways. The analytical framework developed here, centred on molecular characterisation, regulatory pathway clarity, clinical rationale, market size, and milestone proximity, provides a basis for distinguishing structurally sound programmes from those using reformulation framing without the underlying regulatory or clinical logic to support it.
This article is for informational purposes only and should not be considered financial advice. Investors should conduct their own research and consult with financial professionals before making investment decisions. Forward-looking statements regarding clinical timelines and development milestones are subject to change based on manufacturing, regulatory, and operational factors.
Drug reformulation involves converting an existing, approved oral drug into a new delivery format, such as intravenous, rather than synthesising an entirely new molecule. Because the underlying drug's safety profile is already documented, this approach compresses development timelines and reduces the safety unknowns that make traditional early-stage biotech so risky.
The FDA 505(b)(2) pathway allows a drug applicant to reference existing safety and efficacy data for an already-approved molecule, shifting the regulatory burden from proving a drug works from scratch to demonstrating that the new delivery format achieves comparable plasma concentrations without new safety signals. For reformulation companies, this means smaller bridging studies, shorter timelines, and lower capital requirements before reaching a value inflection event.
ICU delirium is a serious acute neurological condition affecting an estimated 30-70% of intensive care patients, increasing mortality and extending hospital stays. Despite its prevalence, no pharmacological therapy is currently approved for delirium in most of the world, forcing clinicians to use off-label antipsychotics and sedatives designed for chronic psychiatric conditions.
Patrys is targeting first-in-human clinical trial entry for its RLS-2201 injectable quetiapine programme in the second half of 2026, with the bridging study expected to run approximately 3-6 months from initiation and completion potentially extending into early 2027.
Investors should assess five key criteria: whether the underlying molecule has an established safety record, whether a clearly abbreviated regulatory pathway such as 505(b)(2) is in place, whether there is a genuine clinical rationale for the new delivery format, whether the addressable market is large enough to attract an acquirer or partner, and how close the company is to its first value-inflection readout relative to its cash runway.



