OncoSil Reaches Final FDA Stage Before US Approval Decision Within 75 Days

OncoSil Medical has received formal communication from the US Food and Drug Administration (FDA) confirming its Humanitarian Device Exemption (HDE) application for the OncoSil™ device has advanced to the final administrative stage before a potential approval decision. The FDA advised on 3 June 2026 that all outstanding questions relating to the treatment of distal cholangiocarcinoma (dCCA) have been satisfactorily addressed, marking the most advanced stage reached by the company’s US regulatory programme to date.
OncoSil reaches final FDA review stage for US market access
The FDA’s correspondence represents a significant regulatory milestone, confirming completion of the substantive review process for OncoSil’s HDE application. The device is designed to treat dCCA, a rare disease affecting fewer than 8,000 individuals annually in the United States.
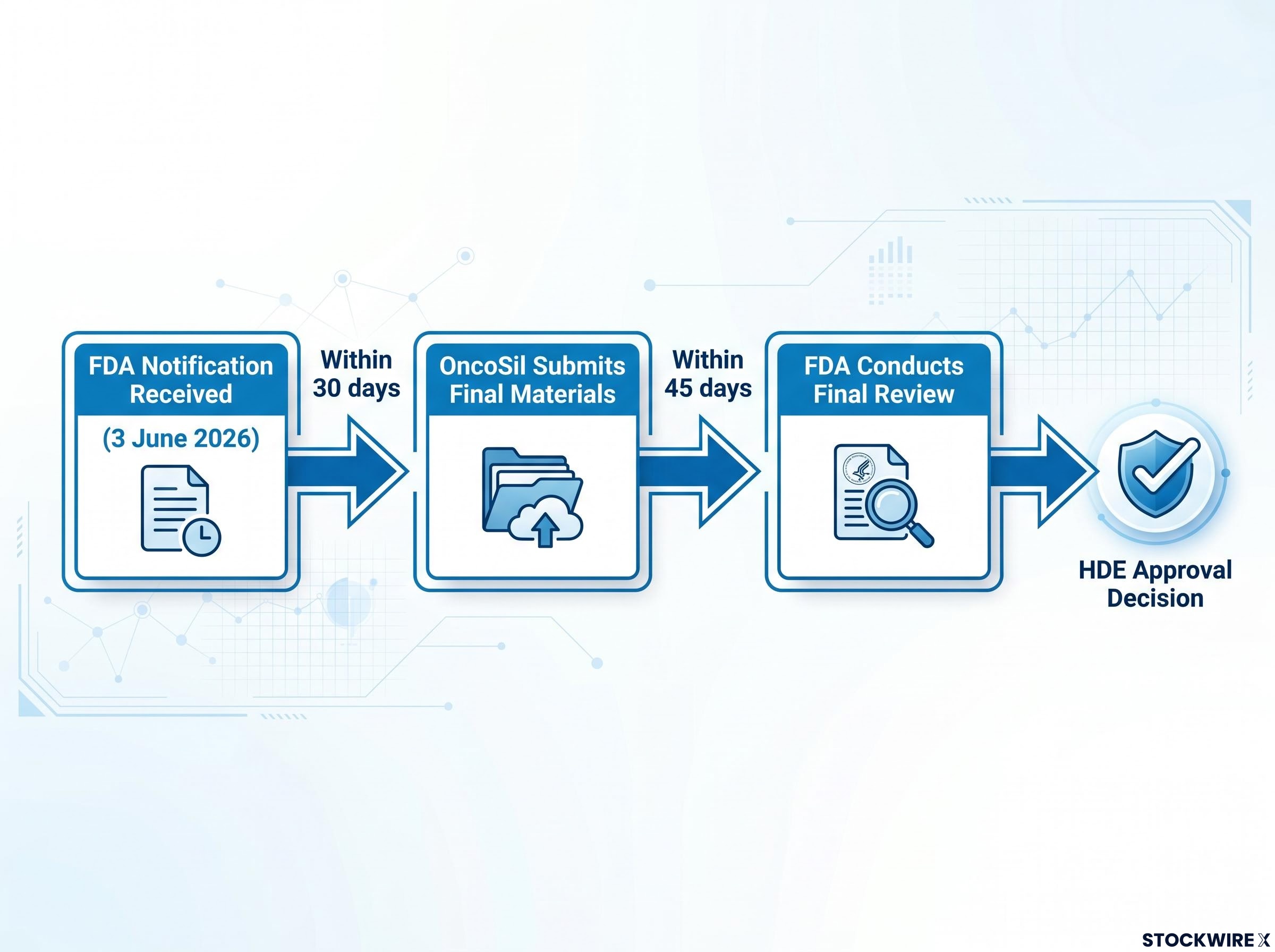
Only two administrative items remain before a potential approval decision. The FDA has requested final device labelling and any modifications to the company’s proposed post-market study, if applicable. OncoSil (ASX: OSL) intends to submit these materials within the FDA’s prescribed 30-day timeframe. Upon receipt, the FDA advised it intends to complete its review within 45 days and grant the HDE.
US market entry represents a transformational commercial opportunity for a company currently generating revenue across 30+ countries but without access to the world’s largest healthcare market. The administrative nature of remaining requirements significantly de-risks the final stage of the regulatory process.
When big ASX news breaks, our subscribers know first
What is a Humanitarian Device Exemption?
The HDE pathway is specifically designed for medical devices targeting rare diseases or conditions affecting fewer than 8,000 individuals annually in the United States. Humanitarian Device Exemption (HDE) is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices — those that support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential unreasonable risk of illness or injury.
Under HDE approval, OncoSil Medical is authorised to commercially market and sell the OncoSil™ device in the US for the specified indication, subject to oversight conditions standard under the framework. The pathway allows the company to generate revenue whilst building evidence for potential broader applications.
The HDE mechanism addresses situations where conventional clinical trial designs cannot be conducted at scale, providing a commercially viable route to market access for devices treating rare conditions with high unmet medical need.
Remaining steps to potential approval
The FDA has confirmed only two items are required before a final decision:
- OncoSil submits final materials (within 30 days from FDA notification)
- FDA conducts final review (within 45 days of submission)
- HDE approval decision
The company intends to submit within the prescribed timeframe and has committed to keeping shareholders informed of material developments. The FDA’s stated intention to grant the HDE upon satisfactory completion provides significant regulatory visibility for investors.
Distal cholangiocarcinoma represents urgent unmet medical need
Distal cholangiocarcinoma is associated with particularly poor clinical outcomes and limited treatment options. Average life expectancy is six months for patients who do not receive treatment for their disease, highlighting the critical gap in therapeutic options for this patient population.
OncoSil’s device addresses this urgent unmet need through targeted intratumoural placement of Phosphorous-32 (32P) microparticles, enabling healthcare professionals to deliver a greater radiation dose directly into the tumour compared to external beam radiotherapy whilst sparing surrounding critical organs.
High unmet need creates favourable adoption dynamics once approved, with physicians seeking any validated treatment option for patients with limited alternatives. The device’s targeted approach differentiates it from conventional external beam radiotherapy by minimising exposure to surrounding tissue.
CEO Nigel Lange commentary
Nigel Lange, CEO & Managing Director
“The FDA’s confirmation that all outstanding questions have been resolved is a major achievement for the Company and an important validation of the extensive work undertaken throughout the HDE review process. Importantly, the FDA has now moved the application to the final stage before potential approval and has requested only the submission of final labelling and any updates to our proposed post-market study. We look forward to completing these final requirements and working with the FDA towards HDE approval.”
OncoSil’s existing commercial footprint
OncoSil™ has received CE Marking approval, providing marketing authorisation in both the EU and the UK. The device holds Breakthrough Device designation in both Europe and the United States, recognising its potential to address unmet medical needs.
Current approvals span 30+ countries including:
TGA approval, secured in May 2026, made OncoSil’s device the first and only Class III medical device approved in Australia for directly targeting pancreatic tumours, adding the company’s home market to its regulatory portfolio ahead of the US decision.
- European Union
- United Kingdom
- Australia
- Türkiye
- Israel
Commercial treatments using the OncoSil™ device have already been undertaken in:
- Spain
- Italy
- Austria
- Germany
- Greece
- Türkiye
- Portugal
- Israel
- United Kingdom
Pancreatic cancer is the 12th most common cancer in men and 11th most common in women globally, with 500,000 new cases detected annually worldwide. Since pancreatic cancer is generally diagnosed at a later stage, it has a poor prognosis for long-term survival.
US HDE approval would add the world’s largest healthcare market to an already established international commercial presence, providing a platform for revenue expansion. The company’s existing footprint demonstrates successful regulatory navigation and commercial execution across multiple jurisdictions.
The next major ASX story will hit our subscribers first
What comes next for OncoSil
OncoSil intends to submit the requested final labelling and any post-market study modifications within 30 days of the FDA’s notification. The FDA has advised it intends to complete its review within 45 days of receiving the submission.
The company held an investor webinar on 9 June 2026 to discuss the significance of the HDE process milestone. Management will continue to keep shareholders informed of material developments throughout the final regulatory stage.
The clear regulatory timeline provides investors with defined near-term catalysts and visibility on potential US market entry timing. Successful HDE approval would represent the culmination of the company’s US regulatory programme and unlock access to a market that has remained outside its commercial reach despite commercial treatments already undertaken across multiple countries in Europe, the Middle East, and Australia.
Investors exploring what additional catalysts may follow the US regulatory outcome should consult our full explainer on the TRIPP-FFX trial and its potential to expand OncoSil’s approved indications, which covers the 88-patient trial design, the late CY2026 regulatory submission timeline, and what a positive readout could mean for the eligible patient base.
Want the Next Biotech Breakthrough in Your Inbox?
Join 20,000+ investors receiving FREE breaking ASX healthcare news delivered within minutes of release, complete with in-depth analysis. Click the “Free Alerts” button at Big News Blast to get real-time alerts the moment market-moving announcements drop, with expert coverage already done for you.