IBM’s Sub-1nm Chip Sends Stock Up 5% in Premarket Trade
32 mins ago

A field that once attracted dozens of competing programmes and hundreds of millions in venture capital has quietly narrowed to a handful of credible contenders. The consolidation of off-the-shelf CAR-T cell therapy development is not a story of failure. It is a story of filtration, one that reveals just how demanding allogeneic cell therapy is to develop, manufacture, and bring through regulatory review.
The premise was always compelling: take CAR-T, the engineered immune cell treatment that has produced durable remissions in blood cancers, and make it available off the shelf from healthy donor cells rather than manufactured individually from each patient’s own blood. That would solve the access bottleneck that limits autologous CAR-T to a fraction of eligible patients. The distance between that premise and clinical reality, however, has been larger than most early investors and developers anticipated.
Here is what the current data from surviving programmes actually shows, what it cannot yet support, and what to watch as the field moves toward the registrational trials that will determine whether the allogeneic promise holds.
The early 2020s saw substantial capital flow into allogeneic CAR-T. By mid-decade, the majority of those programmes have stalled, been deprioritised, or exited entirely. That attrition was not random. Four structural drivers explain why the field contracted:
Manufacturing complexity proved the most decisive filter. Programmes that could not deliver consistent product quality alongside compelling early efficacy and a coherent regulatory plan did not advance. The ones that remain are not simply better funded. They cleared a genuinely demanding technical and strategic threshold.
What defines viability in this narrowed field comes down to four dimensions: manufacturing robustness sufficient for multi-centre trials, clinical evidence spanning more than a single indication, a safety profile that supports outpatient or outpatient-proximate delivery, and regulatory clarity with sufficient capitalisation to reach a registrational trial. Programmes that lack any one of these dimensions face an increasingly difficult path forward.
The phrase “off-the-shelf” suggests simplicity. The engineering reality is the opposite.
Allogeneic CAR-T requires taking T cells from a healthy donor and editing them to accomplish two things simultaneously: prevent the donor cells from attacking the recipient (GVHD prevention, achieved primarily through T-cell receptor disruption) and reduce the recipient’s immune system from rejecting the donor cells (achieved through HLA modulation, where certain surface markers are altered to lower immune recognition). Both steps must be accomplished while preserving the cells’ ability to find and kill cancer.
The core engineering paradox: every edit that makes the donor cells safer for the recipient risks reducing their antitumour potency.
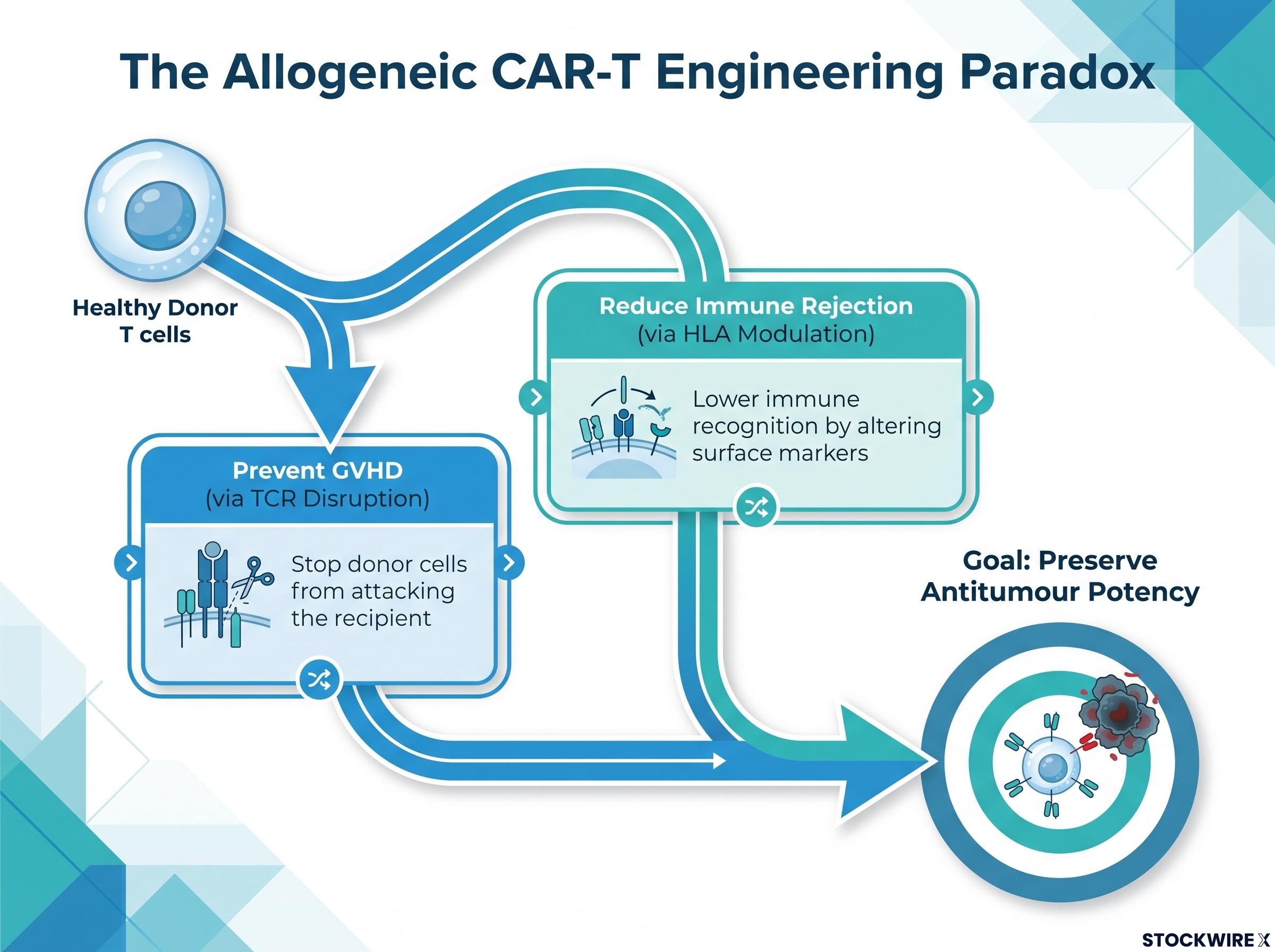
Each additional modification increases process complexity, introduces batch-to-batch variability risk, and raises regulatory questions about whether product lots are sufficiently comparable. These are not abstract concerns. They are the specific reasons most early allogeneic programmes never reached multi-centre clinical testing.
Any programme that has progressed from first-in-human dosing to multi-centre Phase 1b with reproducible product delivery has already passed through the manufacturing filter that eliminated the majority of the field. That context matters when interpreting early-stage clinical data from surviving platforms.
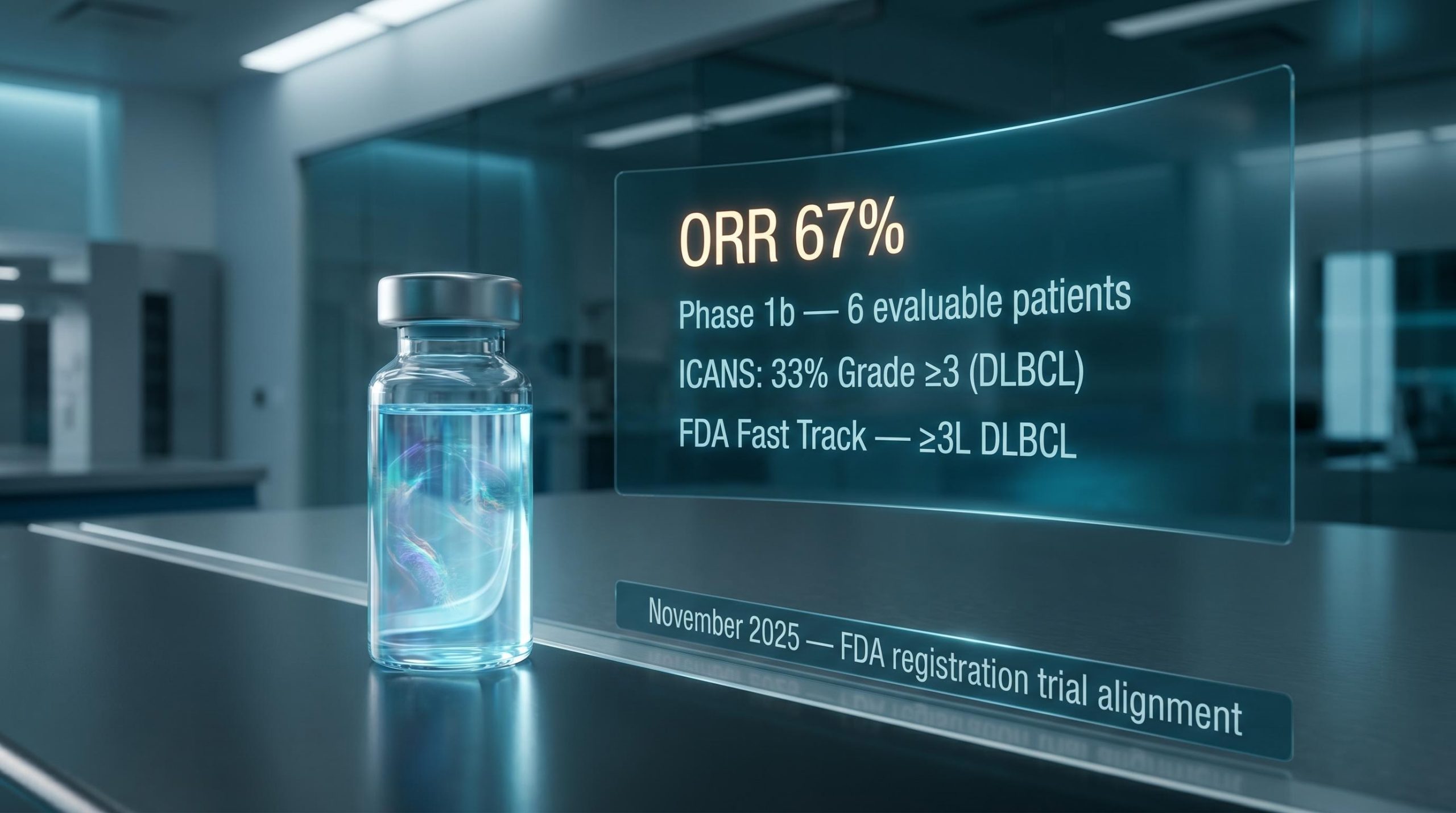
At ASCO 2026, TG Therapeutics and Imugene presented updated Phase 1b findings for azer-cel (azercabtagene zapreleucel), their allogeneic CD19-directed CAR-T therapy. The readout encompassed 24 efficacy-evaluable, CAR-T-naive patients drawn from six distinct B-cell malignancy subtypes, a breadth of indication coverage that is relatively rare for an allogeneic programme at this point in development.
The pre-ASCO efficacy readout published in May 2026, which reported an 81% ORR across 16 evaluable patients, established the baseline signal that the 24-patient ASCO 2026 dataset has since extended and refined across all six B-cell malignancy subtypes.
The aggressive subtype result came first: diffuse large B-cell lymphoma (DLBCL), the most commonly studied CAR-T indication, showed a 67% objective response rate (ORR, the proportion of patients whose disease shrank or disappeared). That is a credible early signal in a heavily pretreated population, though not unprecedented in the CAR-T literature.
The indolent subtype results are where the data becomes more striking.
| Subtype | ORR | Disease category |
|---|---|---|
| DLBCL | 67% | Aggressive |
| MZL (marginal zone lymphoma) | 83% | Indolent |
| CLL (chronic lymphocytic leukaemia) | 100% | Indolent |
| FL (follicular lymphoma) | 100% | Indolent |
| WM (Waldenström macroglobulinemia) | 100% | Indolent |
Combined indolent subtype ORR: 93% across 14 patients
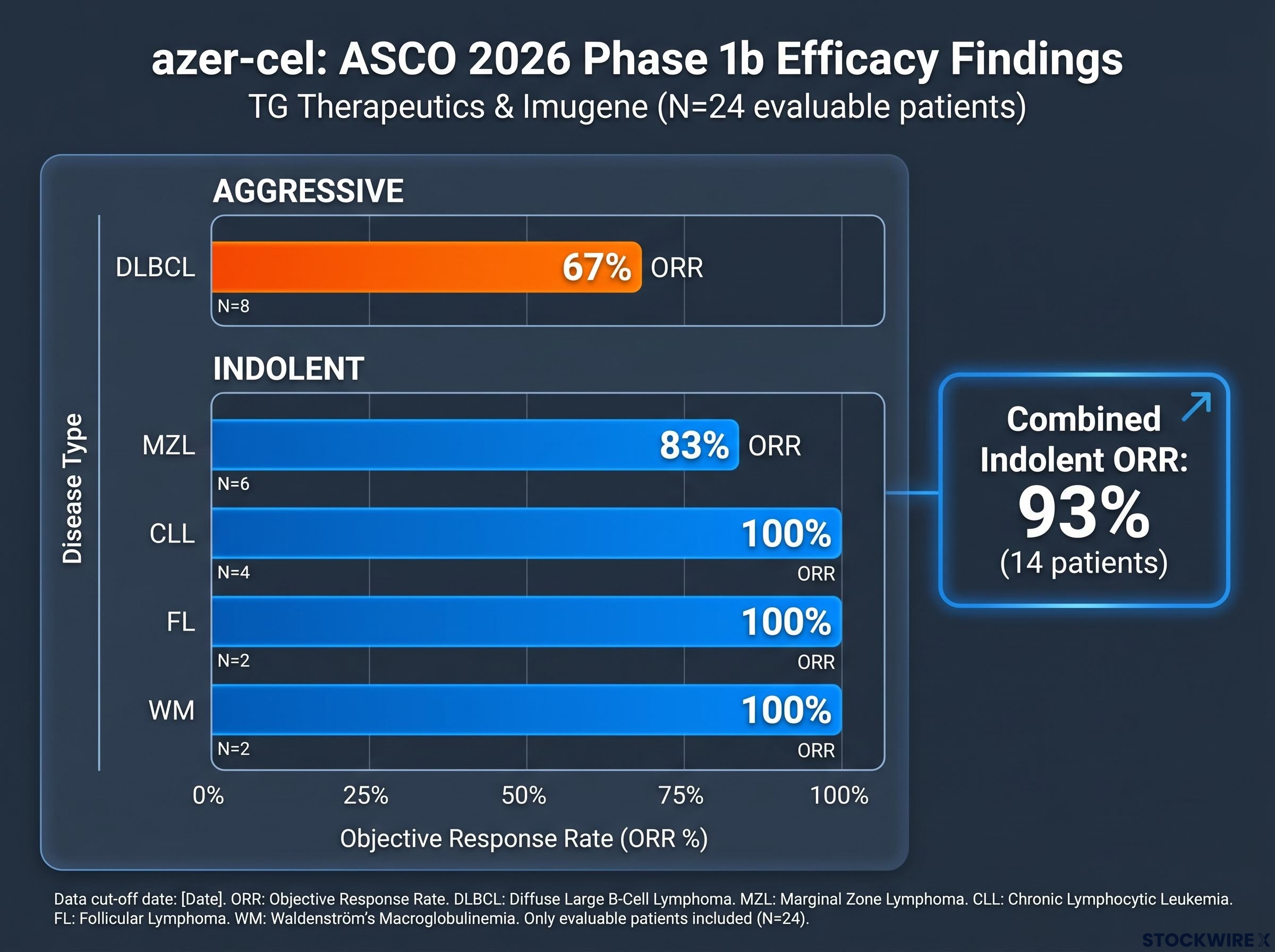
Most allogeneic programmes have concentrated on a single primary indication. Generating evaluable data across six biologically distinct disease entities in one Phase 1b framework is a relative differentiator. The multi-subtype signal matters not because it confirms efficacy, these are small, uncontrolled cohorts that are not predictive of regulatory approval, but because it suggests the platform’s antitumour mechanism is functioning across disease entities with different biological characteristics. That breadth is relevant to how eventual label scope discussions may develop.
The figures above come from small, uncontrolled, early-phase cohorts. They should not be treated as predictive of regulatory approval, commercial success, or future clinical outcomes. Prior results are no guarantee of what follows.
The Phase 1b dataset reported zero Grade 3 or higher cytokine release syndrome (CRS) events as of the ASCO 2026 presentation.
CRS is a systemic inflammatory response triggered when CAR-T cells activate in large numbers. It ranges from flu-like symptoms to life-threatening organ dysfunction. High-grade CRS events have historically been a defining risk of CAR-T therapy, often requiring intensive inpatient monitoring and limiting which patients and which treatment centres can safely deliver the treatment.
CRS severity is measured against the ASTCT consensus grading for cytokine release syndrome, the standardised framework that defines Grade 3 and higher events as those requiring vasopressors or mechanical ventilation, which gives clinical weight to the absence of severe events in the azer-cel Phase 1b dataset.
Key safety observations from the azer-cel dataset:
Lower severe CRS rates in allogeneic settings may reflect differences in how donor-derived cells expand and interact with the recipient’s immune system compared with autologous products. Several allogeneic programmes have reported similar early safety profiles, suggesting this may be a feature of the approach rather than a single programme’s anomaly.
What this means practically: if a product can be delivered with manageable toxicity, the case for outpatient-proximate administration strengthens. That directly addresses one of the access barriers that limits autologous CAR-T, which typically requires specialist inpatient centres with intensive care capabilities. The safety profile does not guarantee outpatient feasibility, but it supports the delivery model that underpins the commercial argument for off-the-shelf cell therapy.
Regulatory alignment is not a formality. In a field where many programmes stalled because they lacked a clear path to approval, formal FDA interactions represent competitive differentiation.
FDA regulatory interactions at this stage of development carry stratified meanings that are not always apparent from press releases alone; a Type C meeting producing confirmed endpoint guidance is categorically different from a Fast Track designation, even though both are routinely reported under similar headline formats.
Azer-cel has secured two regulatory markers:
A Type C meeting is a formal regulatory interaction where the FDA provides structured guidance on development strategy, trial design, and endpoints. Achieving alignment on a single registrational trial, rather than being directed toward multiple studies or additional dose-finding work, provides directional clarity that many competing programmes have not obtained.
Third-line DLBCL patients have progressed through at least two prior lines of therapy. This is a population where autologous CAR-T access is often constrained by manufacturing lead times (the weeks required to produce a patient-specific product), fitness requirements (patients must be well enough to undergo leukapheresis and wait for manufacturing), and treatment urgency in rapidly progressing disease.
An off-the-shelf therapy that can be delivered rapidly to these patients addresses a practical gap, not a theoretical one. The access advantage of allogeneic CAR-T is most concrete precisely where autologous delivery is most difficult.
For the reader, FDA alignment on a single Phase 3 design means the primary remaining development risk has shifted from regulatory uncertainty to clinical execution: can the programme confirm benefit and durability in a randomised setting? That is a materially different risk profile than programmes still negotiating basic regulatory expectations.
These steps do not guarantee approval. Financial projections and clinical development timelines are subject to change based on trial outcomes and regulatory decisions.
The early-stage evidence justifies continued attention. It does not answer the questions that matter most.
T-cell exhaustion, the progressive functional decline of CAR-T cells under sustained antigen pressure, is one of the central reasons durable response in indolent disease remains elusive; the degree to which allogeneic products are structurally less prone to exhaustion than autologous counterparts is an active area of scientific debate with direct implications for how durability data should be interpreted.
The question the field is now asking has shifted. It is no longer “can allogeneic CAR-T generate antitumour activity?” It is “can any remaining programme deliver autologous-like depth and durability of response with superior accessibility and tolerability at commercial scale?”
That question will not be answered until Phase 3 results are available. The early data tells you which programmes are credibly positioned to attempt the answer. It cannot tell you what the answer will be.
Across the four dimensions that define allogeneic viability, manufacturing robustness, clinical evidence scope, safety profile, and regulatory clarity, azer-cel sits among the more advanced CD19-directed programmes still in active, registration-oriented development. Other allogeneic efforts have encountered setbacks, including futility analyses and strategic shifts away from T-cell products, further narrowing the field of credible contenders.
The definitive test remains Phase 3 execution: confirming benefit and durability in a randomised setting against the established autologous standard of care. The specific variables that will determine the outcome, and that you should track in forthcoming readouts, are:
The field has moved past the question of biological plausibility. What remains is the harder question of clinical and commercial proof. The framework above gives you a structured way to interpret the data as it arrives, rather than reacting to headlines without context.
For investors wanting to quantify what the Phase 1b data and regulatory milestones mean for the investment thesis, our deep-dive into azer-cel’s valuation gap examines the peer comparison with Allogene Therapeutics and the specific risk factors, including milestone obligations and dilution exposure, that determine whether the gap narrows.
This article is for informational purposes only and should not be considered financial advice. Investors should conduct their own research and consult with financial professionals before making investment decisions.
Allogeneic CAR T cell therapy uses engineered immune cells derived from a healthy donor rather than the patient's own blood, making it an off-the-shelf product that can be administered rapidly without the weeks-long manufacturing wait required for autologous CAR-T.
Across 24 efficacy-evaluable patients spanning six B-cell malignancy subtypes, azer-cel produced a 67% ORR in DLBCL and a combined 93% ORR across indolent subtypes including 100% response rates in CLL, follicular lymphoma, and Waldenstrom macroglobulinemia, with zero Grade 3 or higher CRS events recorded.
CRS is a systemic inflammatory response triggered when CAR-T cells activate at scale, ranging from flu-like symptoms to life-threatening organ dysfunction; high-grade CRS events have historically required intensive inpatient monitoring, limiting which patients and treatment centres can safely deliver CAR-T therapy.
Azer-cel holds Fast Track designation for relapsed/refractory DLBCL and has reached formal agreement with the FDA through a Type C meeting on a registration strategy built around a single randomised Phase 3 trial in third-line DLBCL, providing directional clarity that many competing allogeneic programmes have not obtained.
Manufacturing complexity was the most decisive filter: each additional engineering step required for GVHD prevention and HLA modulation compounds batch variability and regulatory scrutiny, and programmes that could not deliver consistent product quality alongside compelling efficacy and a coherent regulatory plan did not progress to multi-centre testing.