
Why the US-Iran Deal Won’t Cut Oil Prices Overnight
2 hrs ago

A late-stage metastatic gastrointestinal cancer patient received the lowest tested dose of a CAR T-cell therapy, just 50 million cells, and achieved stable disease lasting 13 months without any further anticancer treatment. That single data point does not prove Chimeric Therapeutics (ASX: CHM) has a commercially viable drug. But it does establish that CHM CDH17, the company’s lead CAR T programme targeting GI cancers, is generating biological activity in patients who have exhausted conventional options. With Phase 1 dose escalation progressing, Phase 2 initiation targeted for mid-2026, and two FDA designations already secured, the programme sits at a genuine inflection point. What follows is an assessment of CHM CDH17 across four layers: clinical evidence, regulatory economics, valuation mechanics, and risk structure. The goal is to equip investors with the vocabulary and framework to form an independent view on whether the current risk-reward profile fits their portfolio construction approach.
CDH17 is expressed on GI tumours, but the protein also carries potential expression on normal tissue. If the engineered T-cells attack healthy cells, the toxicity can be severe and programme-ending. That makes the absence of off-tumour effects the single most important Phase 1 outcome for this construct. It is the binary filter the programme had to pass before anything else mattered.
The data disclosed so far indicate that filter has been passed. At Dose Level 1, Chimeric and its partners reported no safety concerns and no off-target effects. The safety monitoring committee subsequently cleared escalation through Dose Level 2 and into Dose Level 3, with no dose-limiting toxicities (DLTs) at earlier levels. Across the 11 patients enrolled and treated as of April 2026, no off-target tissue toxicity and no severe cytokine release syndrome have been flagged.
Key safety observations from Phase 1 dose escalation to date:
These outcomes are meaningful. They substantially reduce the probability that CHM CDH17 is terminated before reaching a recommended Phase 2 dose (RP2D). They do not, however, confirm safety across all doses and broader patient populations. Higher dose levels may yet surface toxicity signals that earlier cohorts did not. The distinction matters: Phase 1 safety data passing its primary test is not the same as safety being confirmed.
The safety monitoring committee’s clearance of dose escalation to 450 million cells at Dose Level 3 represents the culmination of that sequential de-risking process, with each level providing an independent confirmation that the CDH17 construct does not trigger off-tumour tissue toxicity at progressively higher cell doses.
Under RECIST 1.1, the standard measurement framework used in oncology trials, tumour responses are categorised into a hierarchy. Progressive disease sits at the bottom, meaning the cancer is growing. Stable disease (SD) means tumours have neither grown beyond the progression threshold nor shrunk enough to qualify as a partial response. Partial response (PR) requires at least a 30% decrease in tumour diameter. Complete response (CR) means no detectable disease remains.
| Category | Definition | Threshold | CHM CDH17 Status |
|---|---|---|---|
| Complete Response (CR) | No detectable disease | Disappearance of all target lesions | Not reported |
| Partial Response (PR) | Meaningful tumour shrinkage | ≥30% decrease in tumour diameter | Not reported |
| Stable Disease (SD) | Neither sufficient shrinkage nor sufficient growth | Between PR and PD thresholds | 75% disease control rate (6/8 evaluable) |
| Progressive Disease (PD) | Cancer growing | ≥20% increase in tumour diameter | 2 of 8 evaluable patients |
Stable disease, then, is not a strong efficacy signal in isolation. It sits below partial response in the hierarchy. But context recalibrates the reading entirely. Late-stage, heavily pretreated GI cancer patients frequently progress within months. In that population, disease control from a single infusion at low dose is evidence of genuine biological activity.
The disclosed data show a 75% disease control rate (6 of 8 evaluable patients at 28 days). At Dose Level 2, all 4 patients achieved stable disease by RECIST 1.1. Tumour shrinkage was observed across multiple treated patients.
The most analytically compelling data point is the individual case.
Updated data presented at ASCO in June 2026 extended the programme’s efficacy picture materially: the 82% disease control rate across 11 evaluable patients included CAR T-cell persistence in peripheral blood for up to 15 months post-infusion, a differentiator that addresses one of the field’s core criticisms of solid tumour CAR T durability.
A patient with late-stage metastatic disease, treated at the minimum tested dose of 50 million cells, achieved stable disease lasting 13 months with no further anticancer therapy required during that period.
That figure, from a single patient at the floor dose, does not project into population-level efficacy. The sample remains single-digit, heterogeneous, and uncontrolled. What it does establish is that the CHM CDH17 construct engages its target in vivo and produces durable disease stabilisation in at least some patients. Phase 2 will determine whether that signal translates into a meaningful objective response rate.

CAR T-cell therapy reprogrammes a patient’s own immune cells to identify and destroy cancer. The treatment follows a sequential process:
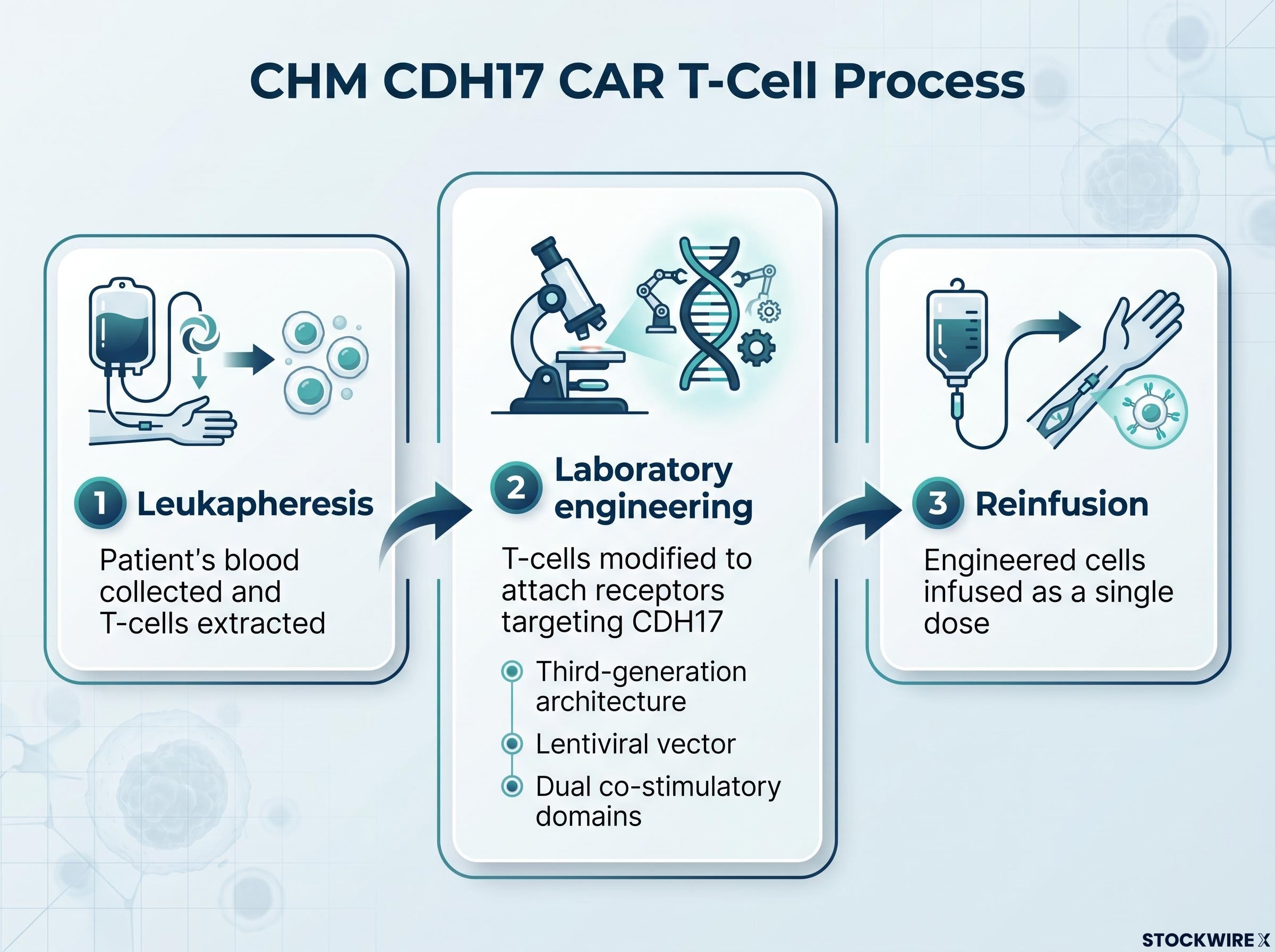
The technology was originally developed at the University of Pennsylvania, the same institution that created Kymriah, the world’s first FDA-approved CAR T therapy. Chimeric’s CHM CDH17 construct is licensed from that programme, providing a direct lineage to the founders of the broader CAR T field.
The first CAR T approvals, Kymriah and Yescarta, targeted blood cancers. Solid tumours present a fundamentally harder problem, and three barriers explain why.
First, the tumour microenvironment in solid cancers is immunosuppressive; it actively disables the immune cells sent to attack it. Second, antigen heterogeneity means not every cancer cell in a solid tumour expresses the same target protein, creating escape routes. Third, autologous manufacturing, where each patient’s therapy is individually produced, carries logistical complexity that scales poorly.
Peer-reviewed research on CAR T barriers in solid tumours identifies the immunosuppressive tumour microenvironment, antigen heterogeneity, and limited T-cell persistence as the three primary mechanisms behind the historically lower response rates seen in solid cancers relative to haematological malignancies, which is precisely the challenge CHM CDH17’s third-generation architecture is designed to address.
CHM CDH17’s construct addresses these challenges through specific design choices: a third-generation architecture, a lentiviral vector, and dual co-stimulatory domains intended to improve T-cell persistence and penetration in solid tumour environments. Whether these features are sufficient is precisely what Phase 2 will test. Against a backdrop of approximately seven decades of conventional chemotherapy as the standard of care, a targeted single-dose cellular therapy represents a materially different therapeutic approach.
GI tumour microenvironment challenges are driving multiple construct-level design responses across the ASX CAR T pipeline; Arovella’s IL-12-TM armouring approach for its CLDN18.2 programme targets the same immunosuppressive tumour environment that CHM CDH17’s dual co-stimulatory domains are engineered to penetrate, with both programmes treating antigen-rich GI malignancies as the test case for next-generation solid tumour cellular therapy.
Both FDA designations improve the project’s financial profile if CHM CDH17 works. Neither moves the needle on whether it will work. That distinction belongs at the centre of any valuation model incorporating these designations.
| Designation | Indication | Key Benefits | Investor Relevance |
|---|---|---|---|
| Fast Track | GEP-NETs | More frequent FDA interaction; eligibility for rolling BLA/NDA review | Reduces regulatory lag, bringing potential revenue forward in DCF models; lowers late-stage design-failure risk |
| Orphan Drug | Gastric cancer | 7-year US market exclusivity; tax credits on clinical expenses; waived or reduced FDA fees | Strengthens post-approval competitive moat; improves project economics; enhances partnering attractiveness |
CHM CDH17’s Orphan Drug Designation provides 7 years of US market exclusivity upon approval for gastric cancer, the most financially concrete economic advantage either designation confers.
Fast Track’s rolling review mechanism allows the FDA to assess sections of a marketing application as they are submitted rather than waiting for a complete filing. In a discounted cash flow framework, this compresses the timeline between trial completion and potential commercialisation. Orphan Drug status delivers direct line-item improvements: tax credits reduce clinical development costs, fee waivers lower filing expenses, and the exclusivity window provides pricing power in a defined market.
The FDA Orphan Drug Designation incentives include seven years of US market exclusivity upon approval, tax credits on qualified clinical trial expenses, and waived user fees, meaning the economic improvements to CHM CDH17’s post-approval scenario are defined by statute rather than dependent on commercial negotiation.
Investors who treat these designations as validation of efficacy will overprice the asset. Those who dismiss them as administrative formalities will underprice the genuine economic improvement they deliver to the post-approval scenario.
In risk-adjusted net present value (rNPV) models, early-stage biotech valuation is driven by the product of two variables: probability of success (PoS) and net present value of peak sales. Because PoS for oncology programmes is lowest at Phase 1 entry and rises materially at Phase 2, even a modest upward revision to PoS at the transition point can reprice equity significantly.
For CHM CDH17, clearing Phase 1 and declaring an RP2D would confirm three things: target biology has passed a real-world test, autologous manufacturing logistics function in practice, and the programme is eligible for Phase 2 design within an accelerated approval context.
Phase 2 will require additional capital. Three financing routes are available, each with distinct implications. An equity raise dilutes existing shareholders. A partnership or licence deal brings non-dilutive capital but shares economics. A strategic transaction may offer validation but cedes control over study design. CAR T precedent suggests larger pharmaceutical partners often seek to participate before Phase 2 begins, positioning themselves to influence the study that could serve as a registration trial. Investors should expect a capital or partnering event around Phase 2 initiation regardless of clinical progress.
The CAR T regulatory precedent amplifies the Phase 2 catalyst. Kymriah and Yescarta were both approved based on single-arm Phase 2 studies without traditional Phase 3 trials. If CHM CDH17’s Phase 2 expansion cohorts deliver a meaningful objective response rate with durable responses and a manageable safety profile, registration based on Phase 2 data is at least conceptually plausible.
The single-arm Phase 2 registration precedent extends across CAR T programmes: Imugene’s azer-cel, which posted an 81% overall response rate across six B-cell malignancy subtypes at ASCO 2026, is similarly targeting a pivotal trial structure that relies on single-arm data, reinforcing that objective response rate and response durability are the metrics regulators have accepted as surrogate endpoints in heavily pretreated oncology populations.
Four catalysts, in likely sequence, define the near-term path:
Each event resets the probability distribution the equity currently prices.
| De-Risked Factors | Outstanding Risks |
|---|---|
| Target biology and early safety (no off-tumour toxicity, no DLTs at low-to-mid doses) | Efficacy magnitude at therapeutic doses in a defined population |
| Manufacturing and logistics feasibility for autologous CAR T | Response durability beyond early stable disease signals |
| Regulatory path economics (Fast Track and Orphan Drug designations) | Competitive positioning against emerging GI immunotherapy approaches |
| Fully funded through Phase 1 completion; lean US-based operational team | Phase 2 financing path (dilution risk or shared economics) |
| Institutional partnerships (Case Western, MD Anderson) co-funding secondary programmes | Single-asset concentration: CHM CDH17 dominates the equity story |
CHM is best viewed as a pre-inflection call option on CHM CDH17’s Phase 2 success. The regulatory designations skew the payoff profile more favourably than a typical early-stage oncology asset, but the underlying binary structure that defines clinical-stage biotech remains fully intact.
Position sizing should reflect the binary nature of the outcome. If Phase 2 delivers a compelling objective response rate with durable responses, the regulatory and commercial infrastructure is already partially built. If efficacy disappoints, the early safety data will not rescue the investment thesis.
Phase 1 has passed its most consequential test. The CHM CDH17 construct has shown no off-tumour toxicity across three dose levels, early efficacy signals indicate genuine biological activity in a patient population with few remaining options, and two FDA designations have improved the programme’s economic profile without altering clinical probability.
The Phase 1-to-Phase 2 transition moves the equity from “concept with early evidence” to “emerging asset with a defined catalyst path.” The RP2D declaration and Phase 2 design announcement are the next specific events that will test whether the repricing implied by that transition is warranted. If Phase 2 data deliver a meaningful objective response rate with durable responses in a defined GI cancer population, the regulatory infrastructure, including a potential path to registration without a traditional Phase 3 trial, is already in place. If efficacy disappoints, the asset is binary and early safety data will not change that outcome.
This article is for informational purposes only and should not be considered financial advice. Investors should conduct their own research and consult with financial professionals before making investment decisions. Past performance does not guarantee future results. Forward-looking statements regarding clinical trial outcomes and regulatory approvals are speculative and subject to change based on clinical developments and market conditions.
CHM CDH17 is Chimeric Therapeutics' lead CAR T-cell therapy programme targeting the CDH17 protein expressed on gastrointestinal tumours, including gastric cancer and gastroenteropancreatic neuroendocrine tumours, designed as a single-dose autologous cellular therapy for patients who have exhausted conventional treatment options.
Across 11 patients treated as of April 2026, Chimeric Therapeutics reported no dose-limiting toxicities at Dose Levels 1 and 2, no off-tumour or off-target tissue toxicity, and no severe cytokine release syndrome, with the safety monitoring committee clearing escalation through three dose levels.
The Orphan Drug Designation for gastric cancer provides seven years of US market exclusivity upon approval, tax credits on clinical trial expenses, and waived FDA fees, while the Fast Track Designation for GEP-NETs enables rolling BLA review, compressing the timeline between trial completion and potential commercialisation in discounted cash flow models.
The four near-term catalysts in likely sequence are the formal recommended Phase 2 dose declaration following Phase 1 completion, the Phase 2 expansion cohort design announcement, a potential partnership or strategic transaction around Phase 2 initiation, and interim Phase 2 data reporting objective response rate and duration of response.
Updated data presented at ASCO in June 2026 showed an 82% disease control rate across 11 evaluable patients, including CAR T-cell persistence in peripheral blood for up to 15 months post-infusion, which is notable given that heavily pretreated late-stage GI cancer patients frequently progress within months on standard approaches.




