Alterity Secures FDA Alignment on Phase 3 Trial for Drug That Slowed MSA 48%

Alterity secures FDA alignment on pivotal Phase 3 trial for ATH434 in MSA
Alterity Therapeutics (ASX: ATH) has achieved a successful End-of-Phase 2 meeting with the US FDA, securing agreement on the key design elements for its registrational Phase 3 programme for ATH434 in Multiple System Atrophy (MSA). This represents a material de-risking milestone that substantially reduces uncertainty around the pivotal programme design for a disease that currently has no approved therapy.
The Phase 3 programme is designed to support a potential New Drug Application in MSA. Trial activities are on track to initiate by year-end 2026.
The FDA concurred with Alterity’s proposed Phase 3 trial design, including the study population, treatment duration, and primary endpoint. Critically, the FDA agreed on the 50mg twice daily dose regimen, which in Phase 2 achieved a 48% slowing of disease progression compared to placebo. Approximately 200 patients will be enrolled in the Phase 3 trial for 12 months treatment duration.
FDA alignment on a Phase 3 design removes a major regulatory uncertainty for late-stage biotech investors and establishes a clear registrational pathway for ATH434.
When big ASX news breaks, our subscribers know first
What the FDA agreed to
The FDA concurred with Alterity’s proposed study population, treatment duration, and primary endpoint for the Phase 3 programme. Significantly, the FDA agreed with the 11-item UMSARS Part I rating scale as the primary endpoint, the same measure where ATH434 demonstrated 48% slowing of disease progression in Phase 2.
The FDA also agreed with the 50mg twice daily dose, which achieved clinically and statistically significant efficacy in the Phase 2 trial. The regulator indicated that Alterity’s planned key secondary endpoints were suitable to support efficacy:
- Swallowing Disturbance Questionnaire
- Orthostatic Hypotension Symptom Assessment
- Clinical Global Impression of Severity
Additionally, the FDA agreed with Alterity’s proposed statistical methods for analysing the primary and key secondary efficacy endpoints. The regulator indicated that the size of the anticipated safety database at the conclusion of Phase 3 was reasonable.
FDA agreement on endpoints and statistical approach signals confidence in the regulatory path forward and validates the Phase 2 clinical strategy.
Why the UMSARS endpoint matters
UMSARS Part I is a functional rating scale that assesses disability in activities of daily living affected by MSA. The 11-item version evaluates orthostatic symptoms, swallowing, speech, handwriting, cutting food, dressing, hygiene, walking, falling, urinary function, and bowel function.
ATH434’s Phase 2 results demonstrated clinically and statistically significant improvement on this exact measure, with 48% slowing of disease progression compared to placebo. The FDA’s acceptance of UMSARS Part I as the Phase 3 primary endpoint validates Alterity’s clinical strategy and creates continuity between the Phase 2 success and the registrational trial design.
Endpoint alignment between Phase 2 success and Phase 3 primary measure strengthens the probability of replicating positive results in the pivotal programme.
Phase 2 data underpinning the Phase 3 design
The Phase 2 ATH434-201 trial was a randomised, double-blind, placebo-controlled investigation evaluating 12 months treatment with ATH434 in patients with MSA. The study enrolled 77 adults who were randomly assigned to receive ATH434 50mg or 75mg twice daily or matching placebo.
The headline efficacy result showed 48% slowing of disease progression versus placebo on modified UMSARS Part I. Additional efficacy assessments demonstrated improvement consistent with the positive UMSARS Part I findings, including the Clinical Global Impression of Severity Scale and patient-reported outcomes assessing swallowing impairment and orthostatic hypotension symptoms.
Biomarker findings revealed that both dose levels reduced iron accumulation in MSA-affected brain regions and demonstrated trends in preservation of brain volume. Wearable sensors indicated that patients receiving ATH434 experienced less decline in outpatient activity levels compared with placebo.
ATH434 was well tolerated with adverse event rates similar to placebo and no serious adverse events attributed to ATH434.
A MuSyCA composite scale analysis from the same Phase 2 dataset showed ATH434’s 50mg dose achieving a statistically significant 41% relative treatment effect versus placebo at Week 52, corroborating the UMSARS Part I findings across an independent composite outcome measure.
| Parameter | Phase 2 | Phase 3 |
|---|---|---|
| Design | Randomised, double-blind, placebo-controlled | Randomised, double-blind, placebo-controlled |
| Patients | 77 | Approximately 200 |
| Treatment duration | 12 months | 12 months |
| Primary endpoint | Modified UMSARS Part I | 11-item UMSARS Part I |
| Dose | 50mg/75mg bid | 50mg bid |
Regulatory designations support expedited development
ATH434 has received Fast Track Designation from the FDA for MSA. The therapy has also received Orphan Drug Designation from both the FDA and the European Commission for MSA.
Fast Track Designation enables more frequent FDA interactions and potential rolling review of regulatory submissions, expediting the development timeline. Orphan Drug Designation provides market exclusivity incentives for rare disease therapies, offering commercial advantages upon approval.
These designations recognise the significant unmet medical need in MSA, where no approved therapies exist to slow disease progression.
The End-of-Phase 2 outcome builds on a sequence of prior regulatory checkpoints, including FDA manufacturing alignment secured in April 2026, when the regulator endorsed ATH434’s chemistry, manufacturing, and control standards for both Phase 3 trial use and potential commercialisation.
Fast Track and Orphan Drug status provide potential commercial advantages including expedited review timelines and market exclusivity upon approval, creating a differentiated competitive position for first-to-market entrants.
Understanding Multiple System Atrophy
MSA is a rare neurodegenerative disease characterised by failure of the autonomic nervous system and impaired movement. The disease causes progressive loss of function and death of different types of nerve cells in the brain and spinal cord, resulting in profound disability.
Key symptoms include slowed movement, rigidity, autonomic dysfunction affecting involuntary functions such as blood pressure maintenance and bladder control, and impaired balance and coordination that predispose patients to falls. The disease pathology involves abnormal accumulation of α-synuclein protein within oligodendrocytes, the support cells of the central nervous system, along with progressive neuronal loss in multiple brain regions.
MSA is a rapidly progressive condition that affects up to 50,000 individuals in the US. Whilst some symptoms can be managed with medications, currently there are no drugs that slow disease progression and there is no cure.
The absence of any approved disease-modifying therapy creates a significant commercial opportunity for first-to-market entrants in a patient population with urgent unmet medical need.
Management perspective
David Stamler, MD, CEO
“Achieving alignment with FDA at the End-of-Phase 2 meeting is a critical step for our Phase 3 programme in MSA, providing the clarity we need to advance to this next stage. We are encouraged by the FDA’s agreement with us on the key elements of our Phase 3 programme, namely the study population, efficacy endpoints, treatment regimen and anticipated safety database, providing a well-defined registrational pathway built on the strength of our Phase 2 data. The successful outcome of the meeting is an important de-risking milestone and gives us confidence as we finalise the protocol and prepare to initiate trial activities by year-end 2026.”
Dr Stamler emphasised that the favourable outcome demonstrates the team’s depth of experience in collaborating with the FDA on neurology development programmes, and expressed confidence that ATH434 is well positioned to become a disease-modifying therapy for individuals living with MSA.
The next major ASX story will hit our subscribers first
Next steps and timeline
The company is finalising the Phase 3 protocol following the successful End-of-Phase 2 meeting with the FDA. Trial activities are on track to initiate by year-end 2026.
The Phase 3 trial will enrol approximately 200 patients who will be randomly assigned in a 1:1 ratio to 12 months treatment with ATH434 50mg twice daily or matching placebo. The primary endpoint will be the 11-item UMSARS Part I, with key secondary endpoints including the Swallowing Disturbance Questionnaire, the Orthostatic Hypotension Symptom Assessment, and the Clinical Global Impression of Severity.
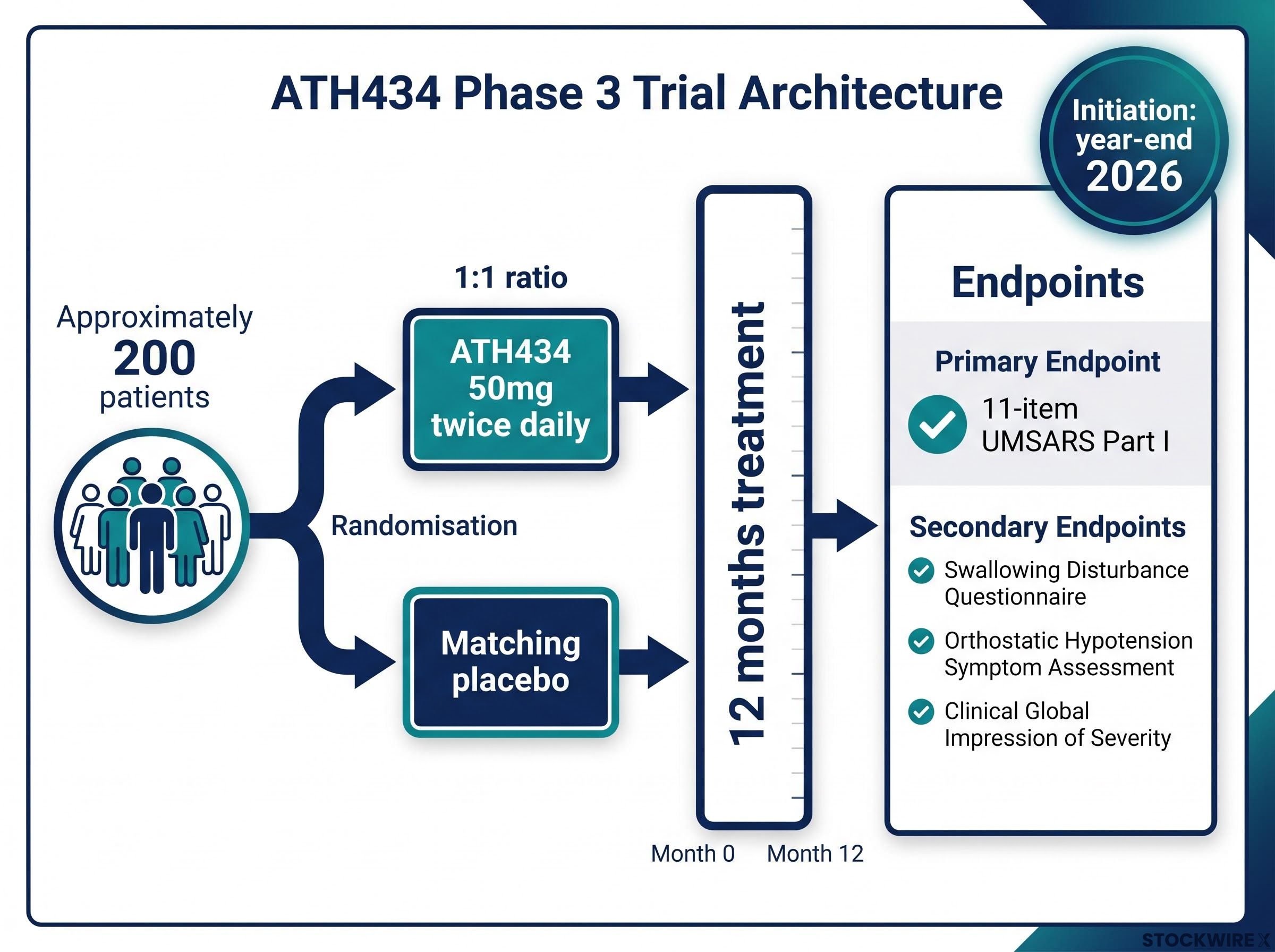
This Phase 3 programme represents the final stage of clinical development required to seek regulatory approval in the United States and is designed to support a potential New Drug Application filing in MSA.
Clear timeline visibility to a pivotal catalyst provides investors with a defined path to value inflection. Phase 3 initiation by end of 2026 sets up potential readout approximately 12+ months thereafter, establishing the regulatory and commercial pathway for a first-in-class therapy in a disease with no approved treatment options.
Don’t Miss the Next Biotech Breakthrough
Join 20,000+ investors receiving FREE ASX healthcare alerts within minutes of release, complete with in-depth analysis on late-stage trials, FDA milestones, and regulatory catalysts. Click the “Free Alerts” button at Big News Blast to get market-moving biotech news delivered straight to your inbox the moment it breaks.